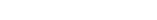仿制药一致性评价一般流程
1 前期调研
1) 企业根据自身情况确定需要做一致性评价的品种和顺序。
2) 确定参比制剂的可及性:如果有参比制剂,参比制剂的选择顺序:国内上市的原研药品、经审核确定的国外原研企业在中国境内生产或经技术转移生产的药品、未进口原研药品。
在原研药品停止生产或因质量等原因所致原研药品不适合作为参比制剂的情况下,可选择在美国、日本或欧盟等管理规范的国家获准上市的国际公认的同种药品、经审核确定的在中国境内生产或经技术转移生产的国际公认的同种药品。源自(关于发布化学仿制药参比制剂遴选与确定程序的公告(2019年第25号))
如果没有参比制剂,需要据情况做临床有效性研究。
2 参比制剂购买
购买前确定参比制剂是否需要备案。取得参比制剂后开始评价。
1) 国外购买:
需申请一次性进口批件,提交以下资料:
1. 进口药品批件申请表及报盘文件
2. 公司合法登记证明文件
3. 申请报告
4. 承诺书
5. 国外获准上市证明
办理进口药品备案和清关,提交以下资料:
1. 进口药品的《进口药品批件》
2. 申请人机构合法登记证明文件复印件
3. 原产地证明复印件
4. 货物合同复印件
5. 装箱单、提运单和货运发票复印件
6. 药品说明书及包装、标签式样
2) 国内购买:首次合作需首营资料。
3 体内体外评价
需确定是否可以豁免生物等效性试验(BE)。
如果可以豁免BE:
1) 企业申请豁免BE,经一致性评价办公室审核通过。
2) 进行全面的体外对比研究。
如果不可以豁免BE:
1) 全面的体外对比研究。
2) 体外一致。
3) 放大试验和BE试验样品制备。
4) BE机构遴选和取得伦理批件。
5) BE试验备案。
6) 进行BE试验。
7) 体内一致。
4 整理申报资料
1) 申报资料报送省级药监局。
2) 药监局进行形式审查。
3) 受理后出具受理通知书。
4) 对研制现场,生产现场进行检查并出具检查报告(60个工作日)。
5) 现场抽取连续生产的3批样品送指定的药品检验机构(30个工作日)。
6) 药检机构出具复核检验报告报送省局。
7) 省局汇总资料报送一致性评价办公室。
8) 一致性评价办公室进行技术审评(60个工作日)。
9) 符合要求,进行公示。
10) 企业取得批件,申报补充申请备案,修改说明书。